🧬 FoldEx
🏆 Winner — Cornell Claude Builder Club Social Impact Hackathon (Spring 2026, Biology & Physical Health track), inspired by Dario Amodei's Machines of Loving Grace.
From "we found a mutation, but we don't know what it means" to a structured, source-cited research report — in minutes instead of weeks.
⚠️ FoldEx is a research-support tool, not a diagnostic device. Every output is meant for review by a qualified clinician or geneticist.
The Problem
When a genetic test returns a variant of uncertain significance (a mutation whose clinical effect is unknown), interpreting it means manually stitching together evidence from a dozen siloed databases, structural prediction tools, and sparse literature — work that can take a specialist days or weeks per variant. FoldEx compresses that workflow into one structured, source-cited report so a human reviewer can focus on judgment instead of plumbing.
What it does
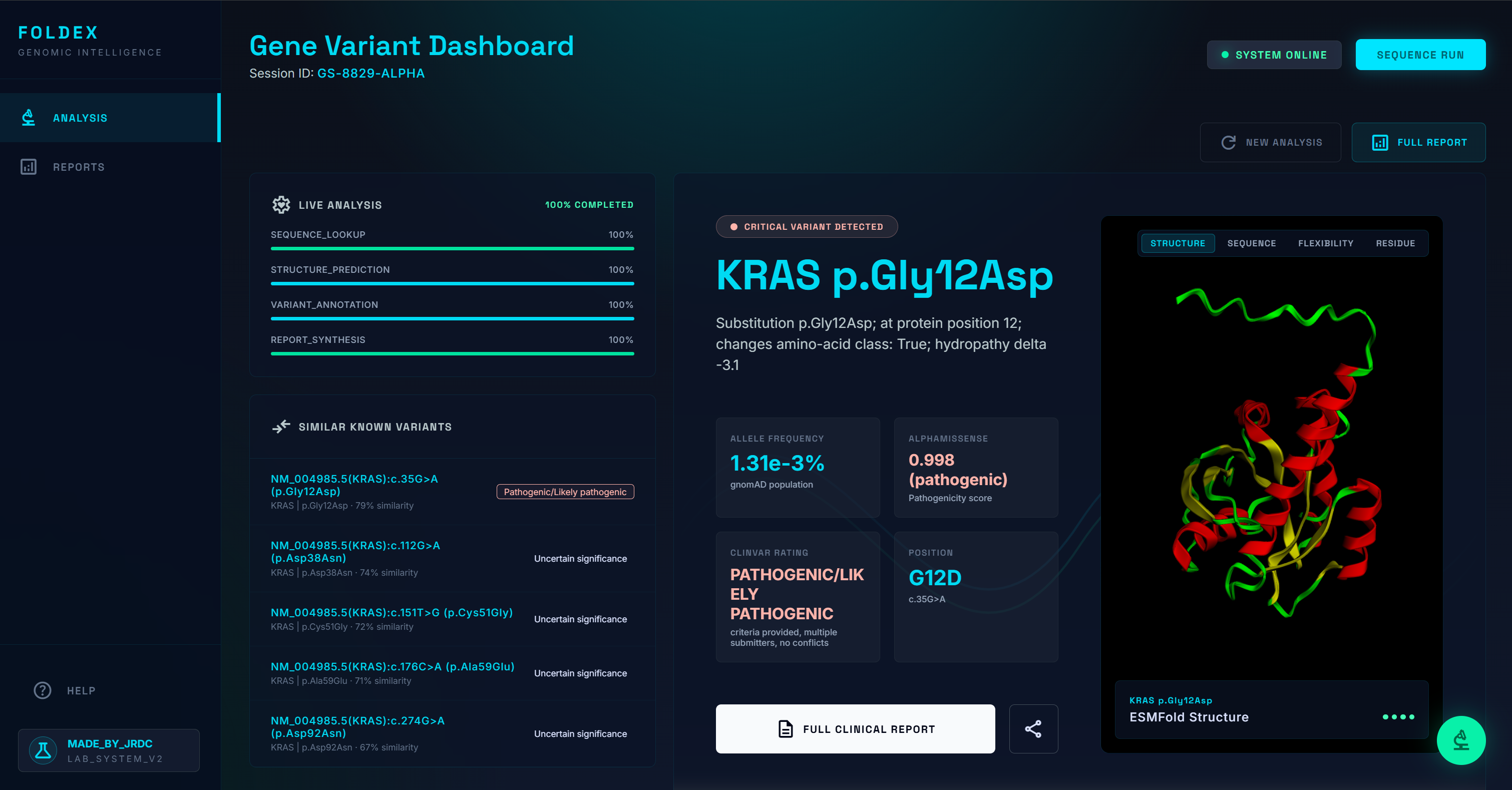
A user submits a single gene variant as text (e.g. BRCA1 c.5096G>A p.Arg1699Gln) or as a lab-report PDF. FoldEx then:
- Parses and normalizes the variant to HGVS standard form using Claude, validated against VariantValidator / Ensembl VEP.
- Annotates with pathogenicity scores (AlphaMissense, SIFT, PolyPhen), clinical assertions (ClinVar), population frequency by ancestry (gnomAD), and protein metadata (UniProt).
- Predicts 3D structures — wild-type from AlphaFold DB, mutant from ESMFold — rendered in an interactive Mol* viewer with the mutated residue highlighted.
- Finds the most biologically similar known variants, ranked by gene/domain match, residue distance, amino-acid property change, and pathogenicity-score similarity.
- Generates a final structured report via Claude, separating established clinical evidence from computational prediction and flagging weak or conflicting findings.
- Adds a patient-friendly summary in plain language, carrying the same non-diagnostic disclaimer.
How Claude is used (and constrained)
Claude acts as a reasoner over evidence retrieved from authoritative sources — never as a generator of variant data or citations. Parsing output that fails normalization is discarded; the reporter may only cite sources present in the evidence dossier; conflicts (e.g. a high AlphaMissense score vs. a benign ClinVar entry) are flagged, not resolved.
Tech Stack
- Frontend: Vite + React + TypeScript + TailwindCSS, Mol* 3D viewer
- Backend: FastAPI (Python 3.11+), RQ workers for slow structure-prediction steps, Docker
- LLM: Claude API (Anthropic) + Groq
- Bioinformatics: Ensembl VEP, AlphaMissense, ClinVar, gnomAD, UniProt, AlphaFold DB, ESMFold, Biopython, VariantValidator
Team
Jayden Lim · Colin Park · Donte Truong · Ricky Ye